[Error rate calculation] quick calculation for PCR and sequencing errors
Summary
ImprovedQ5 for PCR: 8.4*10^-4
Paired-end sequencing: 6.9*10^-5
Previous
Taq for PCR: 0.162
Single read sequencing: 5.2*10^-2
---------Explanations-----------------
PCR error rate:
Q5: https://www.neb.com/products/m0491-q5-high-fidelity-dna-polymerase
~280 lower than Taq
https://www.neb.com/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr
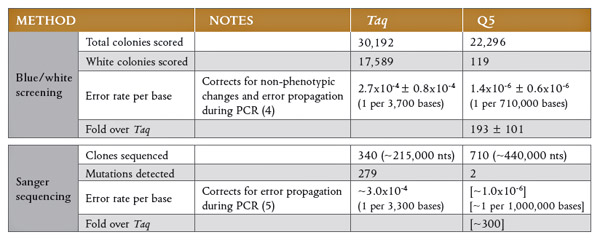
PCR_Q5 = 1.4*10^-6
PCR_Taq =2.7*10^-4
After 30 cycles of PCR amplification for 20bp barcode,
The probability for one of the sites being mutated is
Q5: 1.4*10^-6*20*30 = 0.00084 = 8.4*10^-4
Taq: 2.7*10^-4*20*30 = 0.162
Sequencing error rate:
The HiSeq data sets showed the lowest substitution rates of all three platforms with average rates of 0.0026 (errors per base) for R1 and 0.0040 (errors per base) for R2.
For single read
Error rate = 0.0026*20 = 5.2*10^-2
For paired-end seq, if we threw away reads when there is a base that is inconsistent.
Error occurs only when the base is mutated into the same wrong base
Error rate = 0.0026/3*0.0040/3*3*20 =6.9*10^-5
So by using Q5 and paired-end sequencing, the error rate is still restricted by Q5.
Reducing the number of PCR cycles may help, but not much (linearly), but the product is decreasing exponentially. May reduce a few cycles because over-amplification is not very accurate and may cause additional problems.
If we use 15 cycles
Q5: 1.4*10^-6*20*15 = 0.00042 = 4.2*10^-4
If Q5 doesn't work, Kapa Hifi is another option that works with similar (slightly lower) accuracy.
Reference: Bronner et al, 2009, Improved Protocols for Illumina Sequencing



FYI, with paired-end sequencing of Q5 DNA polymerase, we observed error rates of 1.4 x 10^-3 per 20 bp sequence --just about what should be expected from your calculations.
ReplyDeleteSo it seems like you're right: PCR fidelity still limits the error rate even with Q5. If we want to improve the error rate further, we'd need to reduce the PCR cycles.
Thanks!
I am actually very happy with this. It looks like the two error rates are about the same meaning that we are no wasting effort in either place.
ReplyDelete